
RNA-Seq libraries can be stranded or non-stranded (unstranded). How you prepare the libraries affects the data generated from next generation sequencing (NGS) and its interpretation. In a nutshell, stranded RNA-Seq (also referred to as strand-specific or directional RNA-Seq) enables you to determine the orientation of the transcript, whereas this information is lost in non-stranded, or standard, RNA-Seq. Here, we’ll discuss the methodological differences between the two approaches and how to select the more appropriate one for your NGS project.
How Non-Stranded Library Prep Works
With non-stranded RNA-Seq, you can’t tell whether a sequencing read represents the plus or minus strand of the DNA template. To illustrate why, let’s consider two antisense transcripts originating from the same gene—that is, one mRNA with the plus-strand sequence and the other with the minus-strand sequence. Then, we’ll track how RNA is converted to sequencing products in a non-stranded library preparation (see figure below). For short-read sequencing, the RNA molecules are fragmented after poly(A) selection or ribosomal depletion. Then, random primers are used for first- and second-strand synthesis of cDNA. The ends of the double-stranded cDNA are processed, sequencing adapters are added, and the fragments are amplified. The resulting sequencing products from the two antisense transcripts are identical and can’t be distinguished when sequenced. Thus, information about directionality is lost during cDNA synthesis, which may or may not be needed for your analysis.
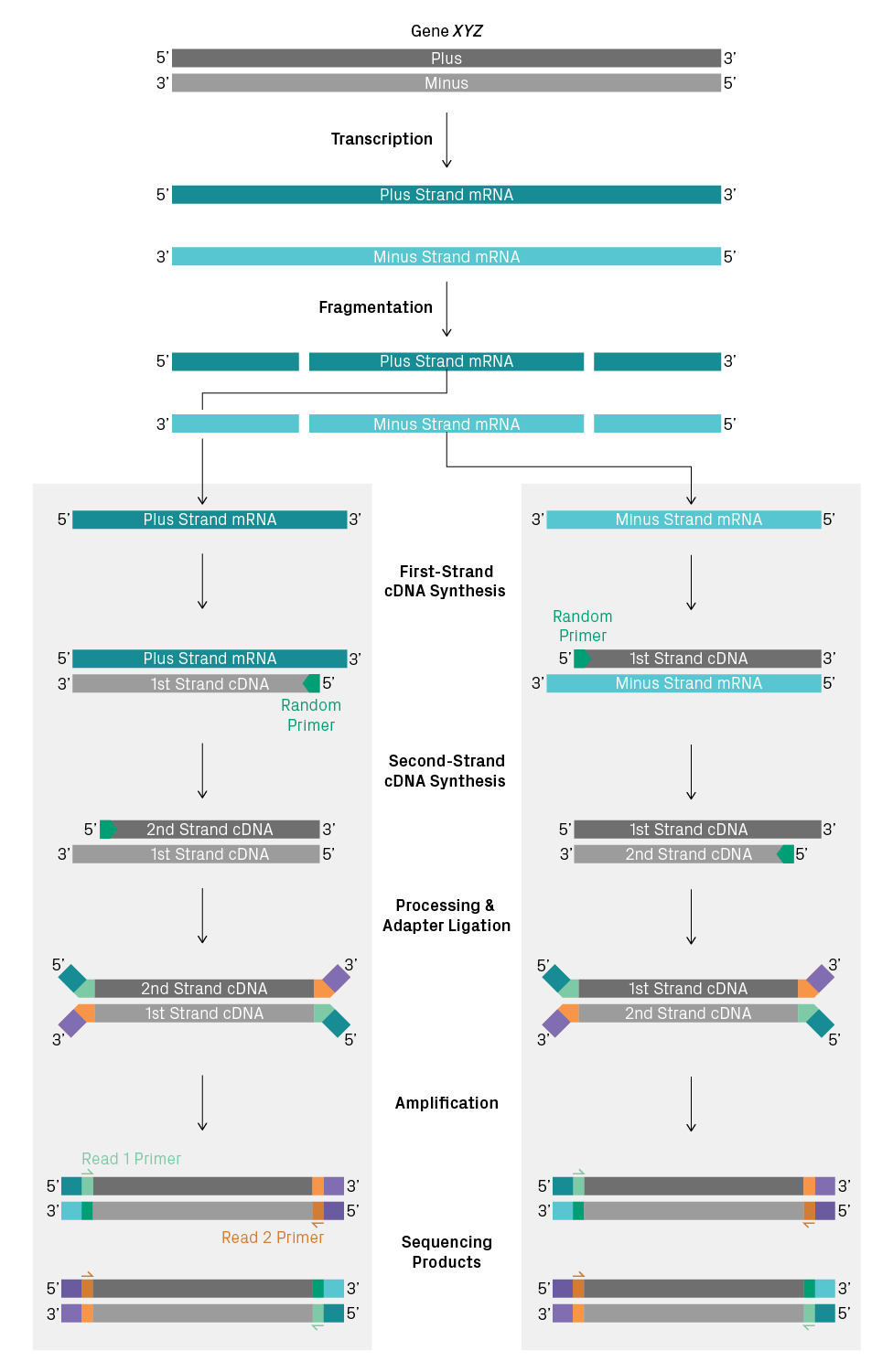
Non-stranded library preparation of two antisense transcripts from the same gene. Information about strand orientation is not preserved during cDNA synthesis. As a result, the sequencing products are identical for the two mRNA molecules, and it’s not possible to determine the directionality of the original transcript directly from the sequencing data.
How Stranded Library Prep Works
In comparison, stranded RNA-Seq distinguishes the first and second strands of cDNA. There are several ways to do this, but, in general, the process is more complicated than non-stranded library prep1. One popular method uses dUTP to label the second strand with uracil instead of thymine (see figure below). After adapter ligation, amplification of the second strand is blocked. This can be accomplished by performing uracil-specific digestion before amplification or by using a DNA polymerase that is unable to amplify uracil-containing templates. As a result, all sequencing products from a particular RNA molecule will have the same orientation between the Read 1 and Read 2 primer binding sites, enabling you to determine the orientation of the transcript.
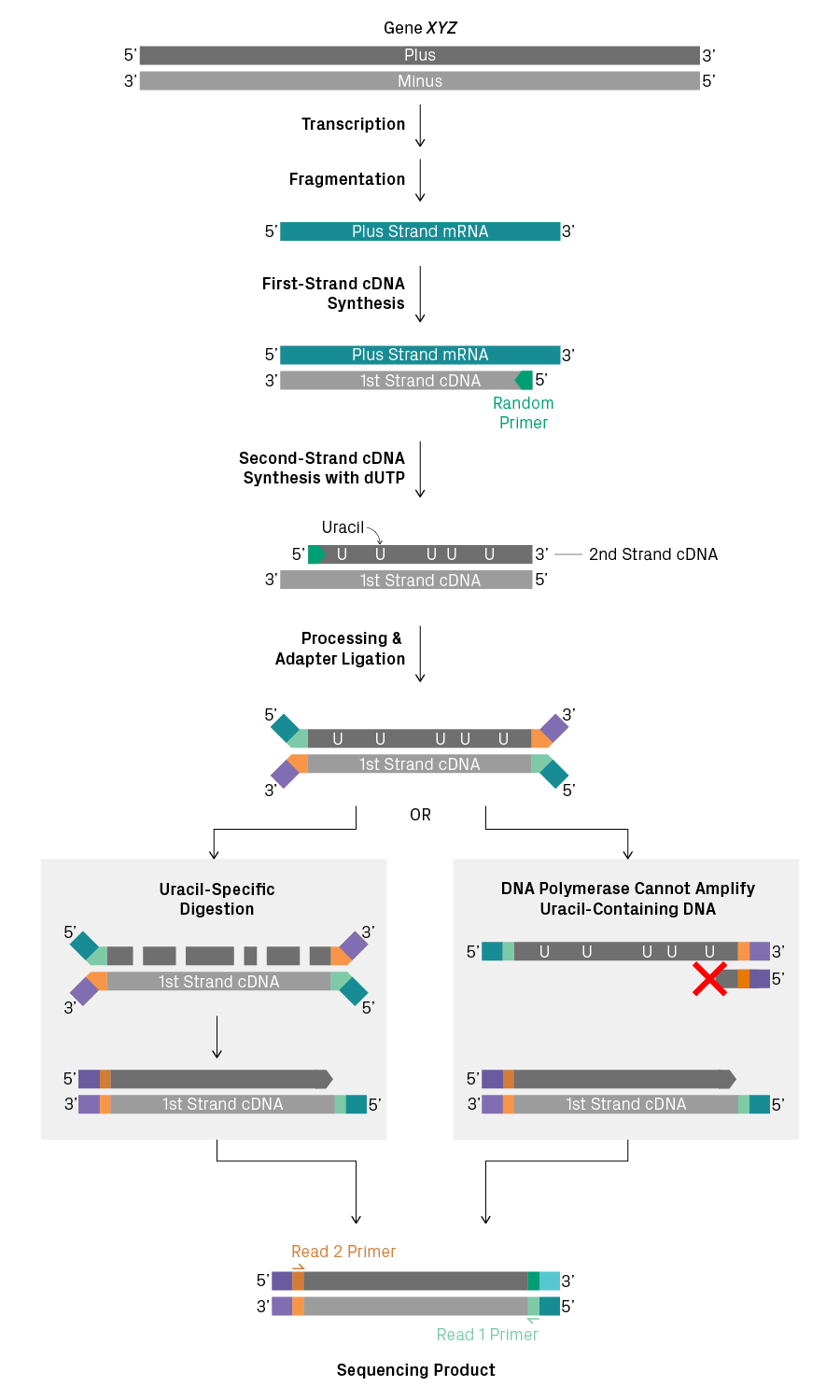
Stranded library preparation using uracil to label the second strand of cDNA. A transcript representing the plus-strand sequence is shown. Amplification of the second strand can be blocked by selective digestion with uracil-DNA glycosylase or by employing a DNA polymerase that can't use uracil as a template, such as Phusion®. The resulting sequencing product has a defined orientation.
Library Preparation Selection Guide
Choosing the right library preparation method depends on several factors, including your experimental objective, budget, and availability of a reference transcriptome for your organism.
The most important consideration is the objective of your experiment. Stranded RNA-Seq is strongly recommended if you’re trying to accomplish one or more of the following, as it’s important to capture information about transcript directionality:
- Identify antisense transcripts
- Annotate a genome
- Discover novel transcripts
Non-stranded RNA-Seq, on the other hand, is often sufficient for measuring gene expression in organisms with well-annotated genomes. With a reference transcriptome, you can infer orientation for most of the sequencing reads. Since there are fewer steps than stranded library prep, the benefits of this approach are lower cost, simpler execution, and greater recovery of material.
Also, when comparing the results of a new experiment to older ones, many researchers prefer using the same RNA-Seq approach. It enables an apples-to-apples comparison of the data.
Key Takeaways
- Stranded RNA-Seq enables you to determine RNA orientation from each sequencing read; this information can’t be directly obtained from non-stranded approaches
- By differentiating the first and second strands of cDNA, stranded library preparation preserves the directionality of the RNA molecule
- Certain applications require a stranded approach; however, non-stranded RNA-Seq is suitable for many NGS projects
Conclusion
Stranded RNA library preparation preserves the directionality of the transcript by distinguishing the first and second strands of cDNA. While most commercial library preparation kits nowadays use a stranded approach, information about transcript orientation may not be needed for your project. Using non-stranded RNA-Seq is generally more economical and less complicated to perform.
For a deeper dive into RNA-Seq, download our eBook, A Guide to RNA-Seq. It covers the what, why, and how of this popular NGS technique and offers valuable information to help you achieve the best results for your RNA-Seq experiment.